药品GMP认证分为国家和省两级进行,根据《中华共和国药品管理法实施条例》的规定,省级以上药品监督管理部门应当按照《药品生产质量管理规范》和药品监督管理部门规定的实施办法和实施步骤,组织对药品生产企业的认证工作;符合《药品生产质量管理规范》的,发给认证证书。其中,生产注射剂、和药品监督管理部门规定的生物制品的药品生产企业的认证工作,由药品监督管理部门负责。
GMP标准(药品生产质量管理规范)是为保证药品在规定的质量下持续生产的体系。它是为把药品生产过程中的不合格的危险降低到小而订立的。
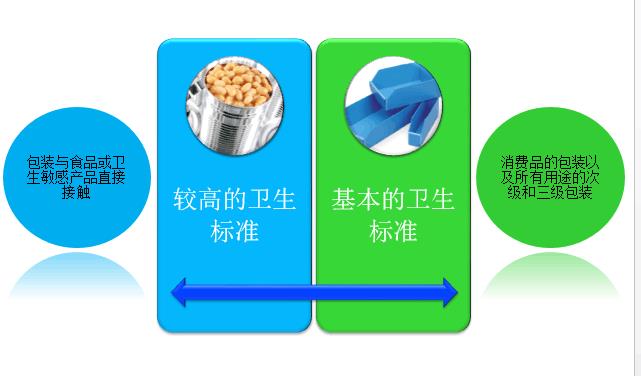
GMP包含方方面面的要求,从厂房到地面、设备、人员和培训、卫生、空气和水的纯化、生产和文件。
“GMP”是英文Good Manufacturing Practice 的缩写,中文的意思是“产品生产质量管理规范”,或是“优良制造标准”,是一种特别注重在生产过程中实施对产品质量与卫生安全的自主性管理制度。

药品GMP认证是国家依法对药品生产企业(车间)和药品品种实施GMP监督检查并取得认可的一种制度。虽然国际上药品的概念包括,但只有中国和澳大利亚等少数几个国家是将人用药GMP和GMP分开的。
GMP认证新版
根据中华共和国部长签署的2011年第79号令,《品生产质量管理规范(2010年修订)》(下称新版GMP)已于2010年10月19日经部务会议审议通过,自2011年3月1日起施行。
新版GMP与98版相比从管理和技术要求上有相当大的进步。特别是对无菌制剂和原料的生产方面提出了很高的要求,新版GMP以欧盟GMP为基础,考虑到国内差距,以WHO2003版为底线。
新版GMP认证有两个时间节点:品生产企业血液制品、疫苗、剂等无菌品的生产,应在2013年12月31日前达到新版品GMP要求;其他类别品的生产均应在2015年12月31日前达到新版品GMP要求。未达到新版品GMP要求的企业(车间),在上述规定期限后不得继续生产品。